
Cours de chromatographie de www.chimie-sup.fr
chapitre I Notions fondamentales
II.2.2 Les colonnes capillaires
II.2.3 Les colonnes semi-capillaires
II.3.1 Caractéristiques d’un détecteur
II.3.4 Détecteur à ionisation de flamme
(FID)
II.3.5 Détecteur à capture d’électrons
II.3.6 Détecteur à photométrie de flamme
(FPD)
II.3.7 Détecteur thermoionique (NPD)
II.3.8 Détecteur à photoionisation
II.4 Classification des phases
stationnaire
II.4.1 Indice de Kovats et droite de
Kovats
II.4.2 Constantes de phases stationnaires
II.5 Optimisation de séparation
chapitre III Chromatographie en phase liquide
III.1 Comparaison chromatographie phase
liquide et phase gaz
III.3 Solvants utilisés en HPLC
III.3.1 Interactions moléculaires entre
phase mobile et solutés
III.3.2 Force éluante et polarité
III.4.1 Dispersion hors colonne
III.4.2 Réservoirs de solvants
III.4.4 Dispositif d’injection
III.5.3 Détecteur fluorimétrique
III.5.4 Détecteur électrochimique
chapitre IV Chromatographie de partage
chapitre V Chromatographie d’adsorption
chapitre VI Chromatographie ionique
VI.2 Chromatographie d’échange d’ions
VI.3.1 Influence de divers paramètres
chapitre VII Electrophorèse capillaire
VII.2.1 Propriétés des électrolytes
VII.2.2 Mobilité électrophoretique µep
VII.2.3 Mobilité électroosmotique µeo
- Coefficient de distribution KA : ![]()
Temps de rétention tR, volume de rétention VR
Temps de rétention tR’, volume de rétention réduit VR’
tR=tR’+t0
t0 : temps mis par la phase mobile pour aller de l’injecteur au détecteur.
- Facteur de capacité k’ : ![]()
Rqe : k’ ne dépend que de la nature des phases.
- Sélectivité α : ![]()
Les pics sont séparés si α>1,1.
Rqe : α ne dépend que de la nature des phases stationnaire et mobile.
- Résolution Rs : ![]()
Les pics sont résolus si Rs>1,5.
Rqe : Rs dépend des conditions opératoires.
Si ω1 = ω2 , ![]() ; En effet si ω1 = ω2 alors
ω1+ω2 = 2ω2 d’où
; En effet si ω1 = ω2 alors
ω1+ω2 = 2ω2 d’où ![]() ,
,
or tR1 = t0(k’1+1) et
tR2 = t0(k’2+1) d’où ![]() , or
, or ![]() ainsi
ainsi ![]()
de plus ![]() soit
soit ![]()
![]()
- Nombre de plateaux N : ![]()
H : hauteur équivalente à un plateau théorique, 100<N<500000.
Rq : Si on augmente la longueur de la colonne, k’ n’est pas modifiée ; le nombre de plateaux ne dépend pas de la nature des phases.
- Nombre de plateaux théoriques : ![]()
N traduit la finesse des pics, il a un effet sur Rs mais pas sur α.
- Dispersion :
Elle est due :
Théorie de Van
Deemter :
H=A+B/u+C.u, approximation car A et C ne sont pas complètement indépendants.
Théorie de Knox :
H=A.u1/3+B/u+C’.u
- Etalonnage externe :
Préparation de solutions de titres connus. Tracé de
Signal=f(C)
- Etalonnage interne :
1. Préparation de solutions étalons A1
de titre x1, A2 de titre x2, A3 de
titre x3. 2. Préparation d’une solution d’étalonnage interne
(EI). 3. Mélange d’un volume constant de A et d’un volume constant d’EI.
V1=VA1+VEI
V2=VA2+VEI
V3=VA3+VEI
Tracé de AA/AEI=f(CA)
Choix de l’étalon interne :
- Méthode des ajouts dosés :
On s’affranchit du volume injecté et de l’ajout d’un composé non présent.
On introduit l’échantillon contenant A et B, et on ajoute des quantités connues mA de A dans l’échantillon.
V1=VAB+VA
V2=VAB+2*VA
V3=VAB+3*VA
Tracé de AA/AB=f(mA)
Interactions :
Soluté Û Phase Stationnaire
Gaz vecteur : H2, He, N2, Ar, CO2 ; le choix du gaz est conditionné par le détecteur.
- L’injecteur amène en tête de colonne l’échantillon sous forme gazeuse avec une dispersion faible, on peut injecter des liquides, des gaz et dans certains cas des solides.
- Mode d’injection :
La phase stationnaire est un solide et met en jeu un équilibre d’adsorption.
o Noir de carbone, graphite (carbopack B) sépare les composés en fonction de la taille.
o Tamis moléculaire au carbone (carbox en 1000) analyse les gaz et les hydrocarbures légers (surface spécifique>1200 m2.g-1).
o Polymères.
o Terre de diatomées (Chromsorb P).
o Silice-Alumine-Zéolithes.
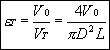
V0 : volume de vide, D : diamètre du tube, L : longueur du tube.
La porosité totale est égale à la somme de la porosité intergranulaire (entre les grains) et intragranulaire (dans les grains).
Si la phase stationnaire est un liquide immobilisé sur un solide, elle met en jeu une constante de partage gaz/phase liquide.
Si la phase stationnaire est une espèce organique liée chimiquement à une surface solide, elle met en jeu une constante de partage gaz/phase greffée.
Exemples de phases
stationnaires :
hydrocarbures ramifiés (squalane), polydialkylsiloxanes (silicones SE30®,
OV17®, CPSIL5®) greffés ou non, polyéthers (Carbowax®) et polyesters (DEGS).
Les colonnes sont constituées d’un tube de silice fondue de diamètre externe 0,25 à 2 mm à l’intérieur duquel est déposée une phase stationnaire de diamètre interne 0,05 à 0,35 mm.
Le volume injecté dans PLOT est plus élevé.
 Sélectivité :
propriété du détecteur à ne pas répondre de la même manière à toutes les
espèces x, capacité à analyser des molécules spécifiques.
Sélectivité :
propriété du détecteur à ne pas répondre de la même manière à toutes les
espèces x, capacité à analyser des molécules spécifiques.
Détecteur à
conductibilité thermique quasi universel comprenant un montage différentiel
(pont de Wheastone) dont les résistances compensent le déséquilibre de
conductivité par une perte de chaleur.
Le gaz vecteur doit avoir une bonne conductibilité thermique tel que H2 ou He et posséder une conductivité différente de celle des solutés.
Caractéristiques :
Détecteur le plus utilisé et spécifique aux composés carbonés sauf HCHO et HCOOH.
Principe :
Caractéristiques :
Détecteur spécifique aux dérivés halogénés. L’électron est formé à partir d’un gaz par des particules β- provenant du 63Ni ou du tritium. N2→N2++e-
Mécanisme : M+e-→M- //
M-+N2+→M+N2
On mesure la variation de courant électrique entre I° (en l’absence de molécules) et I (courant résiduel en présence de soluté). I=I°e-kC , détecteur non linéaire.
Caractéristiques :
Détecteur spécifique du phosphore et du soufre qui émet respectivement à 526 nm et 394 nm.
On utilise une flamme qui brûle les molécules et excite les atomes émettant un rayonnement.
Caractéristiques :
3.10-13 g.s-1 pour P.
Détecteur spécifique aux molécules azotées et phosphorées.
Caractéristiques :
N/P >102.
Les composés à analyser sont ionisés par un faisceau UV intense au Xe ou Ar d’énergie respective 8,3 eV et 11,7 eV ; des électrodes collectent les ions formés.
Applications :
L’objectif est de caractériser la rétention de différents solutés sur une phase stationnaire.
Pour un alcane linéaire : I = 100n, avec n : le nombre d’atomes de carbones.
La droite de Kovats est établie sur une série d’homologues et dans des conditions opératoires fixées en régime isotherme et isobare. Log tR’ = an+b
Pour un composé X : IX = 100α, avec α le nombre fictif d’atomes de carbones de l’alcane linéaire équivalent où
![]()
Il existe des tables de IX afin d’identifier un composé inconnu.
![]()
![]()
Paramètres :
La
température de la colonne suit la loi de Van’t Hoff : ![]()
![]() et par suite
et par suite ![]()
On peut aussi appliquer un gradient de température à la colonne.
Le débit moyen de gaz est donné
par : Dm=0,47 u d
Avec u, vitesse linéaire moyenne du gaz vecteur en cm.s-1 et
d : le diamètre interne de la colonne.
Relation entre Dm et
Rs : ![]()
Interactions :
Soluté Û Phase Stationnaire
Soluté Û Phase Mobile
Avantage CL/CG
Inconvénient CL/CG
![]()
avec
ΔP : perte de charge dans la colonne
Ф : facteur de remplissage de la colonne
η : viscosité dynamique (Pa.s ou Cp)
η(eau) = 0,89 Cp, η(MeOH) = 0,54 Cp, η(ACN) = 0,34 Cp
u : vitesse linéaire moyenne de la phase mobile
dp : diamètre des particules de la phase stationnaire
En HPLC la pression normale est 200 bars ; on peut jouer sur la vitesse u et la viscosité du fluide afin de ne pas dégrader la colonne avec des pressions trop élevées.
Proportions : Tous les solvants existants > solvants chimiquement
compatibles avec l’HPLC > solvants faisant des interactions avec les solutés
> solvants qui vont permettre une bonne séparation.
Différents paramètres
sont à prendre en compte :
Pour les composés polaires : + la phase mobile sera polaire, + elle va entraîner les solutés.
+ la phase mobile sera apolaire, - elle va entraîner les solutés.
Pour les composés peu polaires : + la phase mobile sera polaire, - elle va entraîner les solutés et inversement
On mesure le coefficient de distribution entre le solvant étudié et différentes phases stationnaires
Ex : éthanol : donneur de protons, dioxane : accepteur de protons, nitrométhane : moment dipolaire élevé.
La polarité globale
d’un mélange de trois solvants sera : P’ = logK1+logK2+logK3
En
normant : x1+x2+x3 = 1
Alors
x1 = logK1/P’
Paramètres de
solubilités d’Hidelbrand
![]()
avec Es :
l’énergie moléculaire de cohésion et Vs : le volume molaire de solvant
Paramètres de
solubilités partielles :
δd : interactions de dispersion du nuage électronique, interactions diélectriques
δo : interactions d’orientation, dipôles permanents
δi : interactions dipôles induits
δa, δb : interactions acide/base, par transfert de liaisons H
δtot2
= δd2+δo2+2δoδi+δaδb
|
Phase mobile/soluté |
Non polaire/non polaire |
Polaire/non polaire |
Polaire/polaire |
|
Dispersion |
+ |
+ |
+ |
|
Orientation |
- |
- |
+ |
|
Induction |
- |
- |
+ |
|
Acide/base |
- |
- |
+ |
Cause un élargissement des pics autre que dans la colonne
L’efficacité observée est : l’efficacité de la colonne et la dispersion des solutés hors de la colonne (les tubes de liaisons, volume injecteur, détecteur)
La dispersion due aux tubulures :
On définit L : longueur utilisable pour ne pas augmenter la largeur des pics de 5%
![]()
avec :
Dm : diffusivité du soluté (cm2.g-1)
D : débit d’éluant (mL.s-1)
d : diamètre du tube (cm)
N : nombre de plateaux théoriques
Vr : volume de rétention (mL)
Estimation de la dispersion hors colonne : on enlève la colonne et on injecte les solutés, on obtient ainsi ω(hors colonne) = 4σ(hors colonne)
σtot2 = σcolonne2+ σhors colonne2
Flacons en verre, on utilise une crépine pour éliminer les poussières et les bulles.
1. filtration des solvants
2. dégazages aux ultrasons
Débits utilisés : 0,1-2 mL.min-1
Utilisation de pompes alternative : faible volume interne et utilisable avec des gradients d’élution
On dépose l’échantillon en tête de colonne.
ΔP en HPLC est de l’ordre de qqs dizaines à 100-250 bars.
Vanne d’injection :
6 voies pour les solvants
2 positions load et inject
Tubes en inox de longueur L = 10 à 25 cm
dtube = 4,6 mm le plus courant
dp = 3 à 10 µm
N = 40000 à 60000 plateaux/m
On utilise aussi des micros colonnes
Avec L = 3 à 7,5 cm
Dp = 3 à 5 µm
On limite ainsi la consommation de solvants.
On place souvent des pré-colonnes qui font office de filtres ; elles ont un dp supérieur et protègent la colonne des impuretés.
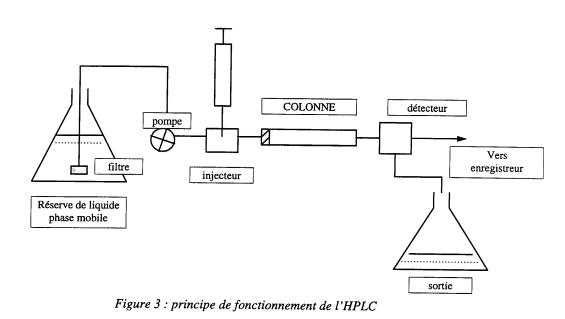
Il mesure l'absorption de la lumière par le produit à la sortie de la colonne. E opère à longueur d'onde constante, celle-ci ayant été fixée par l'opérateur. La lampe Deuterium est utilisé pour des longueurs d'ondes variant de 190-350 nm et la lampe à vapeur de mercure est utilisé à la longueur d'onde non variable de 254 nm. Pour que ce type de détecteur soit utilisable, il faut que :
Les transitions possibles sont :
- π→π* : systèmes conjugués
- σ→σ* : non visible en UV
- n→π* : carbonyles
- n→σ* : alcools
Choix de la longueur d’onde λ de mesure en fonction du soluté et de la phase mobile qui doit peu absorber à cette longueur d’onde.
Loi de Beer-Lambert : Soit un rayon lumineux traversant une solution absorbante de concentration C et de trajet optique égal à l. Si I0 est l'intensité du rayon lumineux à l'entrée de la solution et I son intensité à la sortie, alors :
A = D.O = log (I0 / I) = e l C
où A est l'absorbance, D.O la densité optique, Io l'intensité lumineuse incidente, I l'intensité lumineuse transmise, e le coefficient d'extinction molaire caractéristique de la substance étudiée à une longueur d'onde donnée en L mol-1 cm-1, l l'épaisseur traversée en cm et C la concentration en mol.L-1.
Plusieurs types de détecteurs :
Sur le spectre de soluté :
Il mesure la variation de l'indice de réfraction du liquide à la sortie de la colonne. Cette mesure, extrêmement précise, dépend néanmoins de la température du liquide. On compare cet indice avec celui de la phase mobile pure : il y a donc une référence d'où le terme de variation de l'indice. Ce détecteur exclut les variations de la composition de la phase mobile ; il n'est donc possible de travailler qu'en mode isocratique avec ce détecteur.
Après excitation de l’échantillon dans la gamme UV, on mesure la fluorescence restituée.
Méthode très sensible, qui permet de détecter 1 seule molécule.
Réactions d’oxydoréductions qui produisent un courant proportionnel à la concentration du soluté. Applications : drogues, polluants, produits naturels.
Après nébulisation, évaporation, on envoie un faisceau de lumière - Détecteur universel -
Il faut des différences de diffusivité entre le soluté et la phase mobile.
Interactions : Soluté Û Phase Stationnaire & Soluté
Û
Phase Mobile
La phase stationnaire est soit un liquide immobilisé sur un support par adsorption, soit greffée par liaisons chimiques qui augmente la durée de vie.
Chromatographie de partage à polarité de phases normales : Phase stationnaire polaire, Phase mobile peu polaire.
Chromatographie de partage à polarité de phases inversées : Phase stationnaire apolaire, Phase mobile polaire.
On utilise des ph.
stationnaires polaires : phases greffées (aminopropyl +polaire que
cyanopropyl) et des phases mobiles apolaires pour séparer des composés polaires
et moyennement polaires.
Le pouvoir éluant d’une phase mobile est la somme des polarités de chacun des solvants.
On utilise des phases
stationnaires apolaires : phases C8 ou C18 et des phases mobiles polaires
(eau) pour séparer des composés peu polaires (hydrophobes) et
moyennement polaires.
La phase mobile est constituée d’un fort pourcentage d’eau.
Attention à la miscibilité de tous les solvants ex : MeOH, ACN, THF, CH2Cl2.
Coefficient de partage octanol/eau :
Poe = Kow
= ![]()
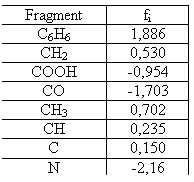
Log Kow = Σfi+cste
Avec, fi
les cstes de Rekker, Log k’ = a log Poe + b, k’ facteur de capacité
et a et b cste dépendant de la phase mobile.
Phases isoéluantes : phases qui conduisent à des k’ voisins mais les interactions mises en jeu peuvent être différentes.
2 phases isoéluantes pour un couple de solutés ne le seront pas pour un autre couple.
loi générale : log k’ = Aφ12+B φ2+C
avec φ1 composition du solvant 1 dans la phase mobile
Pour
un mélange binaire : log k’ = log k’0 – mc
k’0 :
facteur de capacité avec pour ph. mobile (eau)
m :
force de solvant
c :
% de solvant
Pour
un mélange ternaire (eau, solv1, solv2) :
A1φ12+A2φ2+B1Aφ12+B1φ1+B2φ2+C+Dφ1φ2
Pour
une famille d’homologues : log k’ = An+B
Silice : adsorbant très polaire
La capacité d’adsorption et la polarité varient en fonction du taux d’hydratation.
Mélange de solvant
Avec ε0 force éluante
Si ε0 = 0, pas d’adsorption de phase mobile sur phase stationnaire
Si ε0 augmente alors le pouvoir éluant augmente aussi et les molécules de phase mobile prennent la place des molécules de soluté.
La phase stationnaire est immobilisée et la migration se fait par capillarité.
![]()
La vitesse de migration du solvant est ![]() et celle du soluté
et celle du soluté ![]() où tM est
le temps de migration.
où tM est
le temps de migration.
D’où ![]()
Sur la colonne ![]() et
et ![]() d’où
d’où ![]()
Les vitesses étant égales en CCM et en HPLC : ![]()
Loi de rétention : ![]()
Loi de Snyder : ![]()
β* étant le taux d’activité de la silice : si = 1 silice déshydratée, si = 0 silice désactivée.
Le domaine d’application se situe dans la séparation d’isomères.
Elle sépare les solutés ioniques (sels) ou ionisables (molécules organiques) et tend à être remplacée par l’électrophorèse capillaire.
3 types :
La phase stationnaire est ionique :
Une phase stationnaire anionique permet de séparer des cations
La phase mobile est un électrolyte, la rétention dépend du
pH, de la force ionique ![]()
Si I augmente, k’ diminue, il y a rétention de la phase mobile sur la phase stationnaire.
Détection : Conductimétrie : on mesure la conductance de la solution et on place des suppresseurs d’ions pour augmenter la sensibilité.
On ajoute dans la phase mobile un agent d‘appariement d’ions (AAI) = un tensioactif.
La phase stationnaire est similaire à la chromatographie de partage en phase inversée.
Méthode qui permet la séparation d’espèces en milieu biologique (protéines) difficilement séparée par l’HPLC.
L’électrophorèse de zone : migration des espèces chargées dans un champ électrique, et en solution dans l’électrolyte.
Ions, molécules ou particules chargées soumises à un champ électrique subissent une force :
![]()
Force de friction : ![]()
D’où la vitesse de la particule ![]()
Mobilité de l’espèce chargée dans un électrolyte supposé immobile :
Et ![]()
Mobilité de l’électrolyte
Une espèce neutre se déplace à la vitesse ![]()
![]()
Soit la tension appliquée V dans un champ E, avec L, la
longueur du capillaire : ![]()
Soit le temps de migration, avec l, la longueur de migration :
Alors ![]()
Soit qi la quantité de produit détectée ;
alors quelque soit le mode de détection : ![]() , avec Ai la concentration de produit i dans
l’électrolyte et tMi son temps de migration.
, avec Ai la concentration de produit i dans
l’électrolyte et tMi son temps de migration.
En injection hydrodynamique : la composition de l’échantillon n’est pas modifiée.
Alors ![]()
En injection électrocinétique : l’injection se fait via un champ électrique donc les compositions de l’échantillon injecté et de l’échantillon dans l’électrolyte sont différentes.
Alors ![]()
En E.C. les solutés traversent la cellule de détection à leur vitesse propre, ce qui fait qu’on détecte une quantité q de produit dans un volume V de concentration C pendant dt.
Avec un détecteur linéaire la réponse est proportionnelle à la concentration.
Quantité de produit détecté q :
![]()
S : section du capillaire
v : vitesse du produit dans le capillaire
V : volume de produit de concentration C
K : cte